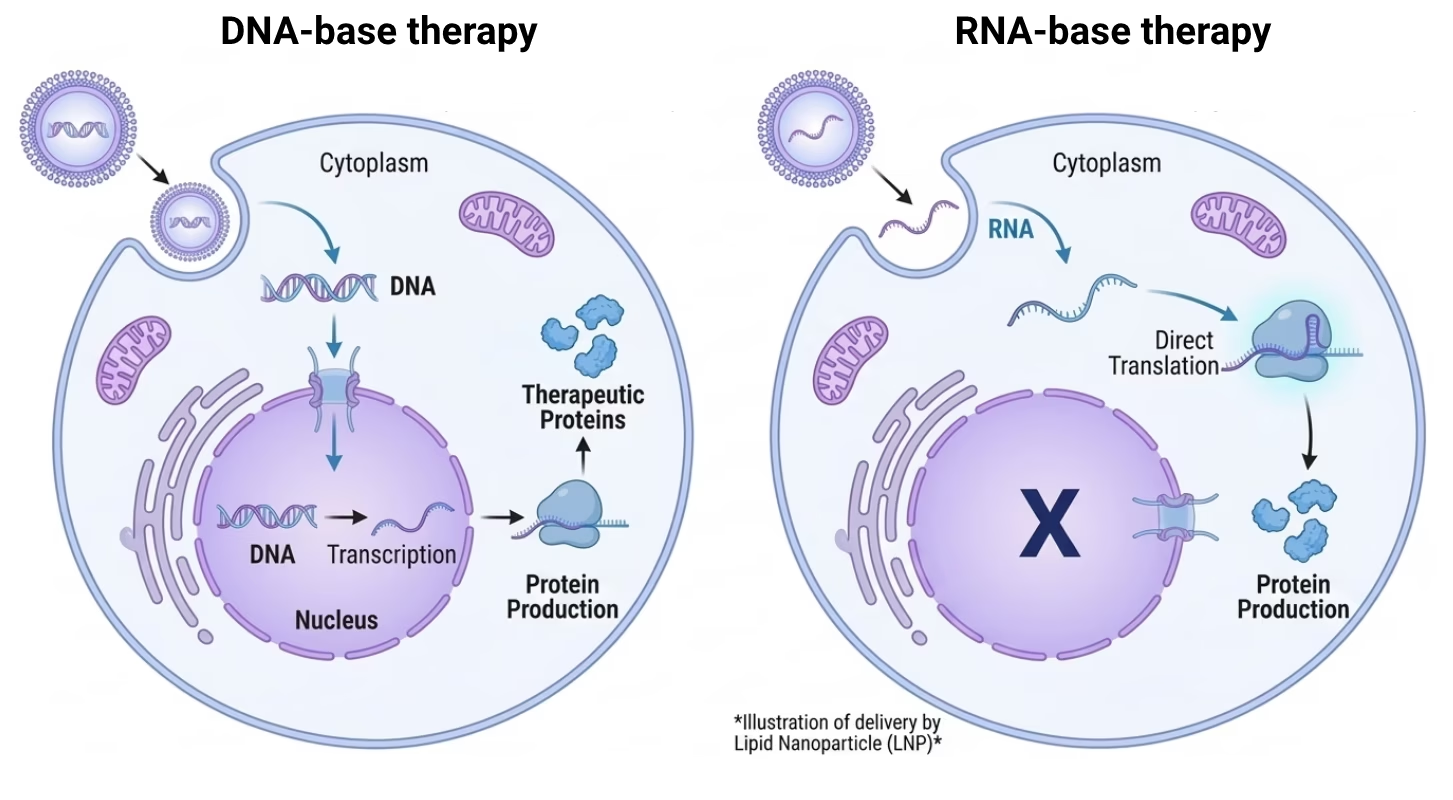
Whether the therapeutic payload is DNA or RNA is one of the earliest and most consequential decisions in a gene therapy program. It determines the intracellular compartment the payload must reach, the delivery platform required, and the downstream implications for expression kinetics, redosing strategy, immunogenicity, and CMC complexity.
DNA requires nuclear entry and depends on mitotic breakdown of the nuclear envelope for efficient translocation, which limits applicability in post-mitotic tissues. RNA acts in the cytosol, bypassing this barrier entirely.
The sections below compare the main viral and non-viral platforms by mechanism, indication fit, and the practical constraints that govern platform selection.
Viral delivery systems for gene therapy
Adeno-associated virus (AAV)
Adeno-associated virus delivers single-stranded DNA that is converted to double-stranded DNA in the nucleus and transcribed by the host cell. It is the most widely used viral vector for in vivo gene therapy.
AAV cannot package RNA: encapsidation requires inverted terminal repeat (ITR) sequences present only in DNA, and RNA lacks the stability to survive AAV production conditions.
Recombinant AAV vectors persist primarily as extrachromosomal episomes in the nucleus rather than integrating into the host genome. This episomal state is stable in post-mitotic cells such as neurons and photoreceptors, which is why AAV has found its strongest clinical footing in non-dividing tissues like the central nervous system and the retina. In dividing cells, episomal DNA is progressively diluted with each round of replication, leading to gradual loss of transgene expression over time.
AAV is therefore well suited to post-mitotic targets and less appropriate where the target cell population turns over rapidly.
However, payload size is limited, redosing is often restricted by neutralizing antibodies, and persistent expression can complicate safety management. Manufacturing scale, cost, and comparability also represent nontrivial constraints.
Lentiviral and retroviral vectors
Lentiviral and retroviral systems integrate DNA into the host genome and are primarily used in ex vivo settings such as cell and gene-modified cell therapies. These platforms enable stable, heritable expression in dividing cells.
Their use is constrained by integration-related safety risks, complex manufacturing, and the permanent nature of genomic modification. As a result, they are generally unsuitable for systemic in vivo delivery.
Non-viral delivery systems for RNA therapeutics
Lipid nanoparticles
Lipid nanoparticles are the most established non-viral delivery platform for RNA therapeutics. They are composed of an ionizable lipid, helper lipids, cholesterol, and a PEG-lipid component. The ionizable lipid remains neutral at physiological pH and becomes positively charged in acidic endosomes, promoting membrane destabilization and cytosolic release.
LNPs are compatible with mRNA, self-amplifying RNA, circular RNA, and siRNA. They protect RNA from degradation and have strong clinical and manufacturing precedent for systemic administration. LNPs have also been used to deliver plasmid DNA in research and early clinical settings, though RNA delivery represents the more clinically advanced application. At the same time, they tend to accumulate in the liver unless specifically engineered, and endosomal escape remains inefficient. Small formulation changes can meaningfully alter biodistribution, immunogenicity, and potency, increasing CMC and comparability risk.
LNP delivery is best understood as a balance between stability, targeting, and intracellular release rather than a fixed solution.
Ligand-conjugated RNA
Ligand conjugation involves chemically attaching a targeting moiety directly to the RNA molecule. The most mature example is GalNAc conjugation, which exploits hepatocyte-specific receptors to drive efficient liver uptake following subcutaneous administration.
This approach offers highly predictable hepatocyte targeting, lower formulation complexity, and durable pharmacodynamic effects that enable infrequent dosing. However, tissue scope is largely restricted to the liver, and the platform is best suited to smaller RNAs such as siRNA or antisense oligonucleotides. It is not a general solution for large transcripts or broad tissue delivery.
DNA conjugation to targeting ligands has been explored, most notably with antibody-DNA conjugates and, in a more applied context, with DNA origami structures functionalized with targeting moieties. However, the clinical maturity is substantially lower than for RNA conjugates, and the approach faces additional constraints specific to DNA: the payload still requires nuclear entry after cellular uptake, which ligand conjugation does nothing to solve. So conjugation can address tissue targeting and cellular uptake, but the intracellular trafficking problem remains.
Polymeric nanoparticles and polyplexes
Polymer-based delivery systems rely on electrostatic complexation between cationic polymers and nucleic acid cargo. These systems can be engineered for degradability, pH responsiveness, and ligand attachment. Polyplexes were originally developed for plasmid DNA delivery and retain applicability for both DNA and RNA payloads, though RNA applications have become the more active area of development.
Their main advantage is chemical tunability, which enables tailored solutions for specific tissues or routes of administration. However, cationic charge is frequently associated with toxicity and immunostimulation, and batch reproducibility can be challenging due to polymer dispersity and self-assembly behavior. Compared with lipid systems, late-stage clinical validation is more limited.
Polymeric systems are powerful but technically demanding and highly sensitive to chemistry and manufacturing control.
Cell-penetrating peptides and peptide-based systems
Cell-penetrating peptides facilitate cellular uptake through membrane interaction or endocytosis and are often combined with motifs intended to enhance endosomal escape. Some peptide systems incorporate nuclear localization signal motifs to support nuclear import of DNA cargo, though this application is less clinically advanced than RNA or oligonucleotide delivery.
These systems are relatively small, modular, and chemically defined, which can be advantageous for local or tissue-restricted delivery. However, in vivo stability can be limited by proteolysis, and cellular uptake does not guarantee productive cytosolic release. Translational consistency varies substantially across tissues and indications.
Peptide-based delivery is best viewed as context-dependent rather than broadly generalizable.
Extracellular vesicles and exosomes
Extracellular vesicles (EVs) are membrane-bound particles secreted by nearly all cell types. They can carry nucleic acids, proteins, and lipids across biological barriers that challenge synthetic systems, including the blood-brain barrier, which has motivated interest in their use as gene therapy delivery vehicles. EVs have been explored for delivery of mRNA, siRNA, miRNA, plasmid DNA, and CRISPR-Cas systems.
Their biomimetic membrane composition may reduce immune recognition relative to synthetic nanoparticles. However, vesicle heterogeneity, limited scalability, and the absence of regulatory-specific technical guidelines for EV-based therapeutics remain significant constraints.
As of 2025, over 290 EV-related clinical trials were registered on ClinicalTrials.gov, though most involve EVs as intrinsic therapeutics based on their endogenous cargo rather than as engineered delivery vehicles of nucleic acids, reflecting the earlier development stage of EV-based gene delivery relative to other non-viral platforms.
Main DNA and RNA delivery methods in gene therapy
The table below compares major gene therapy delivery systems by payload type, durability of effect, redosing feasibility, CMC complexity and typical use case or therapeutic application.
| Delivery system | Payload | Duration | Redosing | CMC Complexity | Use case |
|---|---|---|---|---|---|
|
AAV |
DNA |
Long-term |
Limited by Nab |
High |
In vivo gene replacement, post-mitotic tissues |
|
Lentivirus |
DNA |
Permanent (genomic integration) |
Not needed |
Very high |
Ex vivo cell therapies: CAR-T, HSC editing |
|
LNPs |
RNA (primary), DNA (research) |
Transient |
Yes |
High |
RNA therapeutics, vaccines, gene editing |
|
Ligand-conjugated RNA |
RNA (siRNA, ASO) |
Durable |
Possible, infrequent |
Moderate |
Liver-directed gene silencing |
|
Polymeric nanoparticles |
DNA or RNA |
Transient |
Yes |
Moderate to high |
Research and early-stage; tissue-specific |
Frequently asked questions
What is the mechanistic difference between DNA and RNA delivery in gene therapy?
DNA-based gene therapy requires the payload to reach the nucleus, where it serves as a transcriptional template for the host cell machinery. For plasmid DNA, nuclear entry depends primarily on mitotic breakdown of the nuclear envelope, limiting efficiency in post-mitotic or slowly dividing cells. RNA-based approaches bypass transcription entirely, acting directly in the cytosol. This distinction determines not only the delivery platform required but also expression kinetics, durability, and the intracellular barriers that must be overcome.
Why does AAV persist in post-mitotic cells but lose efficacy in dividing tissues?
Recombinant AAV vectors persist primarily as extrachromosomal episomes rather than integrating into the host genome. In post-mitotic cells such as neurons and hepatocytes, episomal DNA is stable and can support long-term transgene expression. In dividing cells, episomal genomes are not replicated and are progressively diluted with each round of cell division, leading to gradual loss of expression. This makes AAV well suited to non-dividing tissues but a poor choice for applications where the target cell population turns over, such as hematopoietic stem cell modification.
Why does AAV administration generate neutralizing antibodies that prevent redosing, and what strategies are being explored to address this?
Following AAV transduction, capsid proteins are processed through the MHC class I and II antigen presentation pathway, triggering B cell activation and the production of anti-AAV IgG neutralizing antibodies. These antibodies persist long-term and block vector transduction upon re-exposure to the same serotype. Strategies under investigation include capsid engineering to evade existing antibodies, serotype switching, transient immunosuppression prior to redosing, plasmapheresis to deplete circulating antibodies, and IgG-degrading enzymes such as imlifidase. Each approach involves trade-offs in immune burden, manufacturing complexity, and regulatory precedent.
What determines endosomal escape efficiency in LNP-mediated RNA delivery, and why does it remain a bottleneck?
Endosomal escape in LNPs is driven primarily by the ionizable lipid component, which becomes protonated in the acidic endosomal environment and promotes membrane destabilization through formation of non-bilayer lipid structures. However, the efficiency of this process is estimated to be low, with most studies suggesting that only a small fraction of endocytosed LNPs successfully release their cargo into the cytosol. The remainder are trafficked to lysosomes and degraded. Endosomal escape efficiency is sensitive to lipid pKa, LNP composition, particle size, and the rate of endosomal acidification in the target cell type, making it a formulation-dependent variable rather than a solved problem.
What are the CMC implications of switching between RNA payload types within the same LNP platform?
LNP formulations are optimized for specific payload characteristics including molecular weight, secondary structure, and charge density. Switching between payload types, for example from siRNA to mRNA or from mRNA to self-amplifying RNA, requires reassessment of encapsulation efficiency, particle size distribution, polydispersity, and potency. Changes in payload length and structure can alter ionizable lipid to RNA ratios, affect endosomal escape behavior, and shift immunogenicity profiles. Regulatory agencies expect bridging data demonstrating comparability when payload type changes within a platform, and analytical methods validated for one RNA class may not be directly transferable to another.
Why is lentiviral vector manufacturing classified as very high CMC complexity relative to other gene therapy platforms?
Lentiviral vector manufacturing involves transient transfection of producer cells with multiple plasmid components, virus-like particle harvest, and extensive downstream purification to remove process-related impurities including residual plasmid DNA, helper proteins, and empty particles. Lot-to-lot consistency is challenging due to the biological variability of transient production systems. Potency assays must capture both transduction efficiency and integration competence. Long-term follow-up for integration site analysis adds post-manufacturing obligations. Regulatory expectations for comparability, identity, purity, and safety testing are among the most demanding of any gene therapy modality.
What distinguishes GalNAc conjugate pharmacology from LNP-mediated siRNA delivery to the liver?
GalNAc conjugates exploit the asialoglycoprotein receptor (ASGPR), which is highly expressed on hepatocytes and mediates rapid, receptor-dependent endocytosis following subcutaneous administration. After endosomal release, the naked oligonucleotide engages RISC or RNase H depending on the mechanism of action. The result is highly predictable hepatocyte targeting, low formulation complexity, and durable pharmacodynamic effects that enable quarterly or biannual dosing for some approved agents. LNP-mediated siRNA delivery achieves hepatic accumulation primarily through ApoE-mediated uptake following intravenous administration, requires formulation of the oligonucleotide within a particle, and involves endosomal escape as a rate-limiting step. GalNAc conjugates are not a general RNA delivery platform and are unsuitable for large transcripts or extrahepatic targets.
Can lipid nanoparticles deliver plasmid DNA, and what are the key differences from RNA delivery?
LNPs can encapsulate and deliver plasmid DNA, and this application has been explored in research and early clinical settings. However, DNA delivery via LNP introduces an additional intracellular barrier not present for RNA: after endosomal escape, plasmid DNA must cross the nuclear envelope to be transcribed. In non-dividing cells, this requires active nuclear import, which is inefficient for large constructs. Expression onset is also more variable than for mRNA, reflecting the cell-cycle dependence of nuclear entry. As a result, LNP-mediated DNA delivery is substantially less efficient than RNA delivery in most contexts, and RNA represents the more clinically advanced application of the platform.
What are the main analytical challenges in characterizing non-viral RNA delivery systems compared to viral vectors?
Non-viral RNA delivery systems such as LNPs present distinct analytical challenges including measurement of encapsulation efficiency, particle size distribution, polydispersity, surface charge, lipid composition, and RNA integrity simultaneously. Endosomal escape cannot be directly measured in standard release assays and is inferred from potency readouts. For targeted LNPs carrying surface ligands, ligand density, orientation, and binding affinity add further characterization requirements. Viral vectors by contrast have defined capsid structures amenable to established identity and purity assays, though potency and immunogenicity characterization remain complex. The absence of pharmacopoeial standards for LNP characterization means that analytical strategies are largely platform-specific and subject to regulatory negotiation.
Why cannot AAV package RNA?
AAV packaging is dependent on specific DNA recognition sequences called inverted terminal repeats (ITRs), which are located at both ends of the vector genome and are essential for encapsidation by the AAV capsid. RNA lacks ITR sequences and is not recognized by the packaging machinery. RNA is also insufficiently stable under the conditions of AAV production, which involves high temperatures, organic solvents, and density gradient centrifugation steps that would degrade RNA before packaging could occur. These are fundamental biological and biochemical constraints, not engineering limitations that can currently be overcome.
Why does AAV administration prevent redosing with the same serotype?
Following AAV transduction, capsid proteins are processed through the MHC class I and II antigen presentation pathway, triggering B cell activation and sustained production of anti-AAV IgG neutralizing antibodies. These antibodies persist long-term and block vector transduction upon re-exposure to the same serotype, effectively precluding redosing in most systemic indications.
How prevalent is pre-existing AAV seropositivity and what are the clinical implications?
Pre-existing neutralizing antibody seroprevalence ranges from 40% to 70% depending on serotype and geographic region, reflecting widespread prior exposure to wild-type AAV. Seropositive patients are excluded from most systemic AAV trials, as even low-titer neutralizing antibodies can significantly reduce transduction efficiency. This exclusion meaningfully constrains the eligible patient population and complicates patient stratification at the clinical trial design stage.
References
[1] Bhattacharya S, et al. Non-viral Delivery of Nucleic Acids: Insight Into Mechanisms of Overcoming Intracellular Barriers. Frontiers in Pharmacology. 2018. https://www.frontiersin.org/articles/10.3389/fphar.2018.00971/full
[2] Dean DA, et al. Cytoplasmic transport and nuclear import of plasmid DNA. Biochemical Society Transactions. 2017. https://pmc.ncbi.nlm.nih.gov/articles/PMC5705778/
[3] Anzalone AV, et al. Non-viral delivery of CRISPR/Cas system: DNA versus RNA versus RNP. Biomaterials Science. 2022. https://pubs.rsc.org/en/content/articlehtml/2022/bm/d1bm01658j
[4] Naso MF, et al. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs. 2017. https://pmc.ncbi.nlm.nih.gov/articles/PMC5548848/
[5] Hamilton BA and Wright JF. Challenges Posed by Immune Responses to AAV Vectors. Frontiers in Immunology. 2021. https://www.frontiersin.org/articles/10.3389/fimmu.2021.675938/full
[6] Colella P, et al. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Molecular Therapy Methods and Clinical Development. 2018. https://pmc.ncbi.nlm.nih.gov/articles/PMC5758940/
[7] Milone MC and O’Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018. https://www.nature.com/articles/s41375-018-0106-0
[8] Cesana D, et al. Current landscape of vector safety and genotoxicity after hematopoietic stem or immune cell gene therapy. Leukemia. 2025. https://www.nature.com/articles/s41375-025-02585-8
[9] Tanei H, et al. Development of Lipid Nanoparticle Formulation for the Repeated Administration of mRNA Therapeutics. Journal of Controlled Release. 2024. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11109479/
[10] Attia N, et al. mRNA therapeutics beyond vaccines: dosing precision challenges and clinical translation framework. RSC Pharmaceutics. 2025. https://pubs.rsc.org/en/content/articlehtml/2026/pm/d5pm00159e
[11] Huang Y. Preclinical and Clinical Advances of GalNAc-Decorated Nucleic Acid Therapeutics. Molecular Therapy Nucleic Acids. 2022. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8760475/
[12] Gould SJ, et al. Extracellular vesicles: The next generation in gene therapy delivery. Molecular Therapy. 2023. https://www.cell.com/molecular-therapy-family/molecular-therapy/fulltext/S1525-0016(23)00021-7
[13] Chen X, et al. Extracellular vesicle-based drug overview: research landscape, quality control and nonclinical evaluation strategies. Signal Transduction and Targeted Therapy. 2025. https://www.nature.com/articles/s41392-025-02312-w
[14] Luo D and Saltzman WM. Synthetic DNA delivery systems. Nature Biotechnology. 2000. https://www.nature.com/articles/nbt0100_33
[15] Guidotti G, et al. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. British Journal of Pharmacology. 2017. https://pmc.ncbi.nlm.nih.gov/articles/PMC2697800/