This article covers the mechanistic properties, immune considerations, and practical selection criteria for antibodies, aptamers, and peptides as Lipid Nanoparticles (LNP) targeting ligands. Understanding how each ligand class interacts with the LNP formulation itself, not only with the target receptor, is essential for programs moving from in vitro proof-of-concept toward in vivo validation. We hope this will help RNA and gene therapy scientists.
Why ligand selection is a CMC decision, not only a biology decision
When a team decides to surface-functionalize an LNP, the scientific conversation typically begins with receptor biology: which cell expresses the target, at what density, and whether the receptor internalizes productively upon binding. These are necessary starting points, but ligand selection also determines the conjugation chemistry, purification requirements, the analytical panel needed to demonstrate lot-to-lot consistency, the immune activation profile in vivo, and the regulatory characterization burden at IND.
Choosing the wrong ligand class early creates problems that are expensive to fix later, because the choice affects chemistry, analytics, and regulatory strategy, not only biology.
Three examples to illustrate it: a whole antibody conjugated through non-site-specific lysine chemistry produces a heterogeneous population with variable orientation and potential Fc-mediated immune activation. An aptamer attached through a single terminal thiol produces a geometrically uniform surface with tunable density. A peptide conjugated via NHS ester chemistry is synthetically straightforward but may lose activity in plasma within hours due to protease exposure. In each case, the ligand choice directly shapes how the LNP formulation is built, purified, and characterized.
A second critical framing point: the literature on targeted LNPs is almost entirely preclinical, and a large fraction of published data was generated in serum-free or low-serum in vitro conditions that do not reflect in vivo plasma protein environments. Any ligand that works in a buffer does not necessarily work in plasma. This gap between in vitro and in vivo performance is not a failure of any particular ligand class. It is a structural feature of the field that should inform how data are interpreted.
What targeted lipid nanoparticles (tLNP) must accomplish in vivo
A targeting ligand that binds its receptor in a tube must still survive a cascade of biological obstacles before it can deliver cargo to the right cell in a living organism. After intravenous administration, an LNP acquires a protein corona within seconds as plasma proteins adsorb onto the particle surface. This corona can physically occlude surface-conjugated ligands, reducing their receptor accessibility before the particle even reaches the target tissue.
The corona composition is partly determined by the LNP formulation itself, meaning lipid composition, PEG density, and surface charge all influence how well the targeting ligand remains exposed in circulation.
The particle must circulate without immune clearance, extravasate into the target tissue through the local endothelium, contact the target cell, bind the receptor, undergo productive endocytosis rather than surface retention, escape the endosome, and release its cargo into the cytosol.
These requirements explain why single-parameter characterization (binding affinity in buffer, or cell uptake in serum-free media) is insufficient for ligand selection. The relevant characterization set includes performance in physiological protein concentrations, biodistribution in vivo, and immune profile across single and repeat doses.
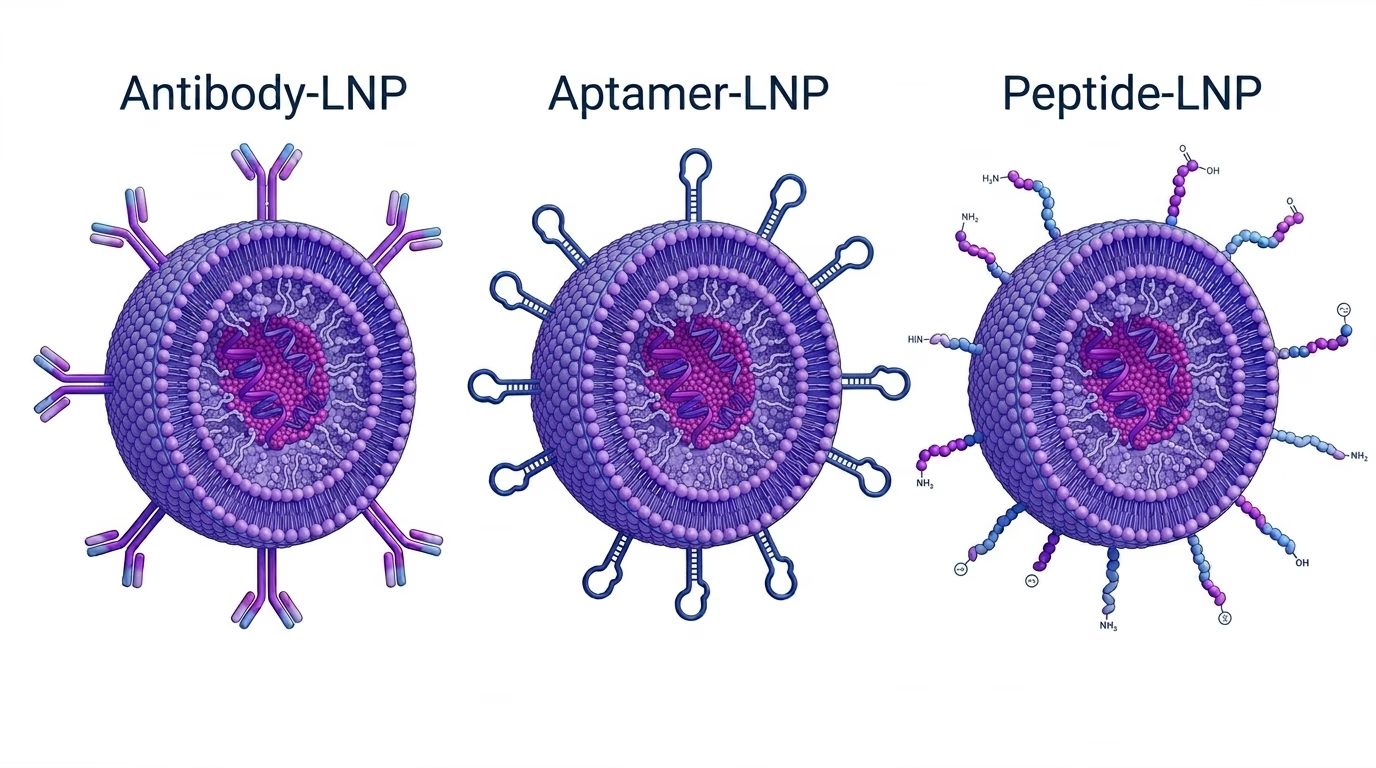
Whole Antibodies
Whole IgG antibodies (~150 kDa) carry the strongest receptor-binding affinity and the deepest clinical history of any targeting ligand class. Dissociation constants in the picomolar to low nanomolar range, bivalent binding through two Fab arms, and broad epitope coverage have made them a natural first choice for proof-of-concept tLNP programs across liver, lung, placenta, immune cell, and tumor targets.
The central liability of whole antibodies on LNPs is the Fc domain. Fc regions engage Fc-gamma receptors (FcgR) expressed on Kupffer cells, splenic macrophages, and neutrophils, driving hepatic clearance through a receptor-mediated route that operates on top of the existing ApoE-LDLR pathway. The practical consequence is that antibody-conjugated LNPs can accumulate more in the liver than untargeted LNPs, even when the intended target is extrahepatic. Fc engagement can also trigger pro-inflammatory cytokine release including IL-6 and TNF-alpha, adding a safety concern on top of the targeting problem.
The large size of whole antibodies limits how many can fit on the LNP surface, reducing the number of opportunities for receptor engagement per particle. The steric bulk of a 150 kDa molecule restricts practical surface density to roughly 5 to 30 antibodies per particle before particle integrity is disrupted. This matters most for cell populations that express the target receptor at low density, where higher ligand density per particle is needed to drive reliable internalization.
Antibody fragments eliminate the Fc domain while preserving antigen binding. F(ab’)2 (~110 kDa) and Fab (~50 kDa) formats both remove the primary driver of FcγR-mediated hepatic clearance and allow modestly higher surface density than intact IgG, at the cost of added manufacturing complexity and lower molecule stability.
In published LNP comparisons, fragment and whole antibody conjugates have shown comparable in vivo targeting performance, with the fragment producing a more favorable immune profile.
Aptamers
Aptamers are short single-stranded oligonucleotides, typically 20 to 80 nucleotides and 1 to 5 kDa, that fold into defined three-dimensional structures capable of binding protein targets with specificity.
Their clinical history as standalone therapeutics is limited but growing: pegaptanib, an RNA aptamer targeting VEGF, was approved for ocular use in 2004, and nucleolin-targeting aptamers such as AS1411 have reached Phase I and II clinical trials in oncology. As LNP surface ligands, they are a more recent application.
In this preclinical project, Shah et al. conjugated a CD4-binding aptamer to SM102-based LNPs and achieved splenic mRNA delivery roughly 80-fold higher than untargeted controls, with targeting efficiency higher to anti-CD4 mAb-LNP and a cleaner cytokine profile than the antibody-conjugated formulation.
Their small size enables display densities in order of magnitude or higher than antibodies. While aptamers generally bind their targets with weaker affinity than antibodies for the same receptor, dense multivalent display on the LNP surface compensates through avidity: multiple simultaneous receptor contacts reduce the effective off-rate, substantially improving apparent binding affinity relative to the free aptamer in solution. This effect is most pronounced for receptors expressed at moderate to high density on the target cell.
Because aptamers carry no Fc domain, immune profile is better and do not activate FcgR-mediated pathways. That way, they avoid the hepatic clearance problem that limits whole antibodies. This is a meaningful advantage for programs requiring repeat dosing, for targets in immune-rich organs where Fc-FcgR interactions would compete directly with the targeting objective, or where cytokine activation is a safety concern.
Unmodified RNA aptamers degrade rapidly in plasma, but standard modifications including 2′-fluoro and 2′-O-methyl substitutions and phosphorothioate backbone changes substantially extend nuclease resistance and are routine practice for therapeutic applications. DNA aptamers are intrinsically more nuclease-resistant and are commonly used in tLNP work with minimal modification.
The historically limiting factor in aptamer development has been the SELEX process, which requires iterative experimental rounds over one to three months with variable success rates and a tendency to enrich sequences with off-target binding if counter-selection is insufficient.
Computational design using machine learning models trained on aptamer-protein binding data is emerging as a faster path, though experimental validation remains required for every computationally generated candidate. The discovery or design timeline should be factored into program planning from the start.
Peptides
Peptides span a broad structural range as LNP targeting ligands. Low molecular weight (0.5 to 3 kDa), synthetic accessibility, low manufacturing cost, and very high achievable display densities on LNP surfaces are their primary advantages.
The central limitation is affinity. Most receptor-targeting peptides operate in the low micromolar to high nanomolar range, substantially weaker than antibodies for the same receptor. High surface density partially compensates through avidity, but competition with endogenous ligands already present in plasma at physiological concentrations further reduces effective receptor binding in vivo. The result is that peptide-targeted LNPs are more susceptible to in vitro-to-in vivo performance gaps than antibody or aptamer systems.
Serum stability is the other major constraint. Linear peptides are substrates for circulating serine proteases, aminopeptidases, and carboxypeptidases. Degradation half-lives for unmodified linear peptides in plasma range from minutes to hours depending on sequence, and cleavage products typically lose receptor-binding activity. Strategies to improve stability include cyclization and modifications, each adding complexity and requiring validation that the binding epitope is preserved.
Differences between targeted LNP types
| Property | Antibody | Ab fragment | Aptamer | Peptide |
|---|---|---|---|---|
|
Molecular Weight |
~150 kDa |
50–110 kDa |
1-5 kDa |
0.5-3 kDa |
|
Receptor Affinity |
pM to low nM |
pM to low nM |
nM to low uM |
nM to uM |
|
Display Density |
Low (5-30) |
Low–moderate |
High (25-100+) |
Very high |
|
Orientation Control |
Poor (non-site-specific) |
Poor–moderate |
Excellent (single thiol) |
Moderate-good |
|
Immune Activation |
Yes ( FcgR binding) |
No (no Fc domain) |
No |
No |
|
Serum Stability |
High |
Moderate-high |
Moderate (modified) |
Low-moderate |
|
Conjugation complexity |
Moderate-high |
High |
Low-moderate |
Low-moderate |
|
Manufacturing cost |
High |
High |
Moderate |
Low |
Antibodies-LNP are more expensive to produce because antibodies require mammalian cell culture, bioreactor infrastructure, and multi-step purification including protein A affinity chromatography. Aptamers and peptides are both produced by solid-phase chemical synthesis, which is faster, more scalable, and does not require biological systems, making them substantially cheaper. Peptides are the least expensive of the three because their synthesis chemistry is simpler and they do not require the nucleotide modifications that aptamers typically need.
Choose ligand based on your LNP program
No single ligand class is best across all target and development contexts. The goal is to match the choice to the real constraints of the program.
Use whole antibodies when: receptor affinity is paramount and high-affinity antibodies are commercially available, proof-of-concept speed matters more than immune profile optimization, the target tissue does not overlap with high-FcgR-expressing macrophage populations, and the program can absorb the analytical complexity of conjugation efficiency characterization.
Consider fragment antibodies when the whole antibody format has confirmed targeting activity but Fc-mediated hepatic clearance or cytokine activation is emerging as a liability. Fragment generation adds an enzymatic cleavage step, increases manufacturing complexity, and requires isotype-matched optimization, so this transition is most justified after proof-of-concept with whole IgG is established rather than at the outset.
Use aptamers when: immune activation must be minimized (repeated dosing programs, immune-cell targeting where Fc-FcgR interference is a direct competitive concern), high surface display density is required for productive endocytosis in a low-receptor-density cell population, manufacturing cost and timeline matter, or the target cell type is in an immune-rich organ such as spleen or lymph node where Fc-mediated hepatic clearance is especially problematic. The SELEX or AI-design timeline should be factored into program planning.
Consider peptides when: the target receptor has a known, high-affinity peptide binder, the delivery route is local rather than systemic (reducing serum stability requirements), or very early discovery-stage screening across many candidate ligands is needed before committing to a more complex ligand class.
For all classes: validate targeting performance in the presence of plasma proteins before reporting in vivo claims. Serum-free in vitro data overestimate selectivity. The protein corona is not optional.
Note: The comparison above applies to systemic intravenous administration of LNPs in preclinical models. Ligand performance under intramuscular, intrathecal, or intraperitoneal routes will differ and requires separate characterization.
How Crystal NAX supports targeted LNP formulation
Crystal NAX provides LNP formulation, conjugation, and analytical characterization support for RNA-LNP programs from discovery through IND-enabling stages. The tLNP service platform covers all three ligand classes discussed in this article.
The tLNP analytical panel goes beyond standard LNP characterization to address the conjugation-specific attributes that carry the highest risk for lot-to-lot variability. It includes physicochemical attributes (DLS, PDI, zeta potential), mRNA content and integrity (UPLC, CE/AGE), lipid content and purity (UPLC-CAD), and tLNP-specific methods: total and conjugated ligand content by HPLC, ELISA, or LC-MS, degree of labeling, conjugation efficiency, and in vitro targeting activity by flow cytometry.
Frequently asked questions
What is the difference between a targeted LNP and a conventional LNP?
A conventional LNP delivers RNA to the liver by default, driven by apolipoprotein E adsorption onto the particle surface and subsequent recognition by LDL receptors on hepatocytes. A targeted LNP engineers an additional layer of selectivity on top of that baseline, either by attaching a receptor-binding ligand to the particle surface to drive active uptake in a specific cell type, or by modifying lipid composition to shift organ-level biodistribution. The key distinction is that targeted LNPs require explicit validation that the targeting mechanism is functioning in vivo, because adding a ligand does not automatically override the default hepatic tropism.
Why does antibody conjugation sometimes increase liver accumulation instead of reducing it?
Whole antibodies carry an Fc domain that engages Fc-gamma receptors expressed on Kupffer cells and splenic macrophages. This creates a new hepatic clearance route that operates on top of the existing ApoE-LDLR pathway, meaning the total liver signal can be higher for antibody-conjugated LNPs than for untargeted ones, even when the intended target is extrahepatic. Antibody fragments such as F(ab’)2 and Fab remove the Fc domain and avoid this problem, at the cost of added manufacturing complexity.
Can aptamers match antibody targeting performance despite weaker individual binding affinity?
Individual aptamers typically bind their targets with nanomolar to low micromolar affinity, weaker than antibodies for the same receptor. When aptamers are displayed at high density on the LNP surface, however, multiple simultaneous receptor contacts reduce the effective off-rate, improving apparent binding affinity substantially relative to the free aptamer in solution. In some preclinical models, aptamer-conjugated LNPs have achieved targeting efficiency comparable to antibody-conjugated counterparts while producing a cleaner immune profile, though this is context-dependent and cannot be assumed across all targets and formulations.
Why do peptide-targeted LNPs often perform better in vitro than in vivo?
Two mechanisms explain most of the gap. First, in vitro assays are typically run in serum-free or low-serum conditions, so the protein corona does not form and the peptide ligand is fully accessible to the receptor. In vivo, adsorbed plasma proteins can partially occlude the ligand before the particle reaches the target tissue. Second, linear peptides are degraded by circulating proteases with half-lives ranging from minutes to hours, meaning the ligand may no longer be intact by the time the LNP extravasates into the target tissue. Both effects reduce in vivo selectivity relative to what in vitro data predict.
What does the protein corona have to do with ligand selection?
The protein corona is the layer of plasma proteins that adsorbs onto the LNP surface within seconds of intravenous administration. For targeted LNPs, the corona can physically occlude surface-conjugated ligands, reducing their receptor accessibility in vivo. This effect applies to all three ligand classes but is particularly important for antibodies because their large size relative to the PEG layer makes them more susceptible to being buried. Ligand accessibility should be validated under physiological protein concentrations before in vivo targeting claims are made, because serum-free in vitro conditions do not capture this effect.
How do I know which conjugation chemistry to use for my ligand?
The choice depends on the functional groups available on the ligand and on the reactive lipids incorporated into the LNP formulation. Maleimide-thiol chemistry is appropriate for thiol-bearing ligands including reduced antibodies and thiol-modified aptamers, and uses DSPE-PEG-maleimide in the LNP formulation. SPAAC chemistry using DBCO-labeled ligands reacting with azide-LNPs produces a more chemically stable bond and avoids maleimide hydrolysis during dialysis, making it increasingly preferred for antibody and aptamer conjugation. NHS ester chemistry is suitable for amine-bearing ligands including peptides. In all cases, the degree of labeling on the ligand and the molar ratio of reactive lipid in the formulation must be optimized, because both affect conjugation efficiency and particle integrity.
Is any of these three ligand classes clinically validated for LNP targeting?
No. As of early 2026, none of the three ligand classes is clinically validated as an LNP surface targeting approach. Aptamers have clinical precedent as standalone therapeutics, with pegaptanib approved for ocular use and several others reaching early clinical trials, but their use as LNP surface ligands remains preclinical. Antibody-drug conjugates and antibody-nanoparticle systems have been explored in oncology clinical trials with mixed results, but the specific combination of antibody conjugation to ionizable LNPs for RNA delivery has not advanced through late-stage trials. Published clinical outcomes with targeted nanoparticles broadly have been mixed, and the in vivo-to-clinical translation gap should be treated as real and unresolved.
When does surface display density matter more than individual ligand affinity?
Display density becomes the dominant variable when the target cell expresses the receptor at low surface density, because a higher number of ligands per particle increases the probability that at least one productive receptor contact occurs during the brief window of cell-particle proximity. It also matters when the ligand has moderate affinity but fast off-rate, because multivalent display reduces the effective off-rate through avidity. Conversely, when receptor density on the target cell is high and the ligand affinity is already in the picomolar range, adding more ligands per particle produces diminishing returns and may introduce steric problems that reduce rather than improve targeting efficiency.
Why is ligand orientation on the LNP surface important and how does it differ across ligand classes?
A ligand that is attached through its receptor-binding domain rather than away from it cannot engage the receptor productively, even if it is present on the surface. For whole antibodies conjugated through non-site-specific lysine modification, orientation varies across the particle population: some molecules present their Fab arms outward and are functional, others face inward and are not. Aptamers conjugated through a single defined 5′ thiol are uniformly oriented, with the binding domain consistently presented outward. Peptides fall in between, depending on where the conjugation site is placed relative to the binding epitope. Poor orientation effectively reduces functional ligand density below what total conjugation efficiency measurements suggest, which is why orientation control matters independently of how many ligands are attached.
What safety readouts should be included in a preclinical tLNP study?
At minimum: organ-to-body weight ratios for liver and spleen, serum pro-inflammatory cytokines including IL-6 and TNF-alpha, and alanine aminotransferase as a liver injury marker. For antibody-conjugated LNPs, Fc-mediated immune markers deserve particular attention because Fc-FcgR interactions can elevate cytokines independently of the RNA cargo. For programs planning repeat dosing, anti-PEG antibody titers and complement activation markers including C3a and C5a should be added, because accelerated blood clearance on subsequent doses can substantially alter the pharmacokinetic and efficacy profile relative to single-dose data.
How does the cost of aptamer or peptide synthesis compare to antibody production at early development scale?
Antibodies require mammalian cell culture, bioreactor infrastructure, and multi-step purification including protein A affinity chromatography, making them the most expensive option at any scale. Aptamers and peptides are both produced by solid-phase chemical synthesis, which does not require biological systems and scales more predictably. Peptides are the least expensive of the three because their synthesis chemistry is simpler and they do not require the nucleotide phosphoramidite reagents and nuclease-resistance modifications that aptamers typically need. At early development scale where many candidate ligands may need to be screened, the cost difference between peptides or aptamers and antibodies can be substantial enough to influence which ligand class is evaluated first.
At what stage of a program should I commit to a specific ligand class?
Ligand class selection ideally happens before formulation development begins, because the choice determines which reactive lipids go into the LNP, which conjugation chemistry is developed, and which analytical methods need to be built. In practice, many programs start with whole antibodies for proof-of-concept because high-affinity binders are commercially available and the biology can be established quickly, then revisit the ligand class decision when immune profile, manufacturing homogeneity, or repeat-dose pharmacokinetics become priorities. The most expensive scenario is completing formulation and process development for one ligand class and then switching, because conjugation chemistry, purification, and analytical methods may not transfer across classes without significant redevelopment.
References
Monopoli MP et al. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nanotechnol. 2012; 7:779-786.
Tenzer S et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol. 2013; 8:772-781.
Geisler HC, Battistini E, Thatte AS, Padilla MS, Mitchell MJ. Preparation of targeted lipid nanoparticles for precision nucleic acid delivery. Nat Protoc. 2026. https://doi.org/10.1038/s41596-025-01330-w
Parhiz H et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J Control Release. 2018; 291:106-115.
Billingsley MM et al. In vivo mRNA CAR T cell engineering via targeted ionizable lipid nanoparticles with extrahepatic tropism. Small. 2024; 20:e2304378.
Ben Mkaddem S, Benhamou M, Monteiro RC. Understanding Fc receptor involvement in inflammatory diseases: from mechanisms to new therapeutic tools. Front Immunol. 2019; 10:811.
Nabih NW et al. Antibody-functionalized lipid nanocarriers for RNA-based cancer gene therapy: advances and challenges in targeted delivery. Nanoscale Adv. 2025; 7:5905-5931.
Nimjee SM, White RR, Becker RC, Sullenger BA. Aptamers as therapeutics. Annu Rev Pharmacol Toxicol. 2017; 57:61-79.
Rosenberg JE et al. A phase II trial of AS1411 in metastatic renal cell carcinoma. Invest New Drugs. 2014; 32:178-187.
Shah S, Ranasinghe M, Decker J et al. Lipid nanoparticles with aptamers enable targeted mRNA delivery to CD4+ T cells. Drug Delivery. 2026; 33(1):2637266.
Menon I et al. Fabrication of active targeting lipid nanoparticles: challenges and perspectives. Mater Today Adv. 2022; 16:100299.
Liao H et al. Strategies for organ-targeted mRNA delivery by lipid nanoparticles. WIREs Nanomed Nanobiotechnol. 2024; 16:e2004.
Kratschmer C, Levy M. Effect of chemical modifications on aptamer stability in serum. Nucleic Acid Ther. 2017; 27:335-344.
Kohlberger M, Gadermaier G. SELEX: critical factors and optimization strategies for successful aptamer selection. Biotechnol Appl Biochem. 2022; 69:1771-1792.
Patel S et al. AptaBLE: A Deep Learning Platform for SELEX Optimization. 2024. https://doi.org/10.64898/2026.01.06.698056
Zhao Z, Ukidve A, Kim J, Mitragotri S. Targeting strategies for tissue-specific drug delivery. Cell. 2020; 181:151-167.
Diao L, Meibohm B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin Pharmacokinet. 2013; 52:855-868.
Vlieghe P et al. Synthetic therapeutic peptides: science and market. Drug Discov Today. 2010; 15:40-56.
Liu Z et al. Engineered multi-domain lipid nanoparticles for targeted delivery. Chem Soc Rev. 2025; 54:5961-5994.